Three-channel analysis tutorial
This tutorial walks through a complete three-channel CellColoc workflow based on the interactive Jupyter notebook
user_scripts/nb_microglia_3D_three_channel_user_script.ipynb,
which is identical to the interactive Python script
user_scripts/microglia_3D_three_channel_user_script.py.
The goal is to show how to analyze a 3D microscopy dataset in which a third channel is not merely displayed for orientation, but is actively segmented and included in the downstream analysis.
In particular, this tutorial demonstrates how to:
segment channel 0, channel 1, and channel 2,
evaluate channel-0 versus channel-1 positivity,
evaluate channel-0 versus channel-2 positivity,
derive channel-0 cells that are positive for both channel 1 and channel 2,
and inspect each of these positivity views separately in napari.
Dataset used in this tutorial
The tutorial uses the microglia example data set distributed with CellColoc in
example_data/microglia_3D/
Please download the example data from the CellColoc Zenodo example-data record first, as described in the Example data set section. Store the downloaded files locally in a convenient place. For the remainder of this tutorial, we assume that the downloaded files are available relative to the current working directory or current example script in:
example_data/microglia_3D/
The script is written to handle one selected file from that folder at a time.
This is a 3D multichannel fluorescence dataset. In this tutorial, we treat:
channel 0 as the primary
cellchannel (Cx3cr1-tdTomatomicroglia reporter signal),channel 1 as the first marker channel (
Iba1staining),channel 2 as the second marker-like channel (
DAPIin this demo setup).
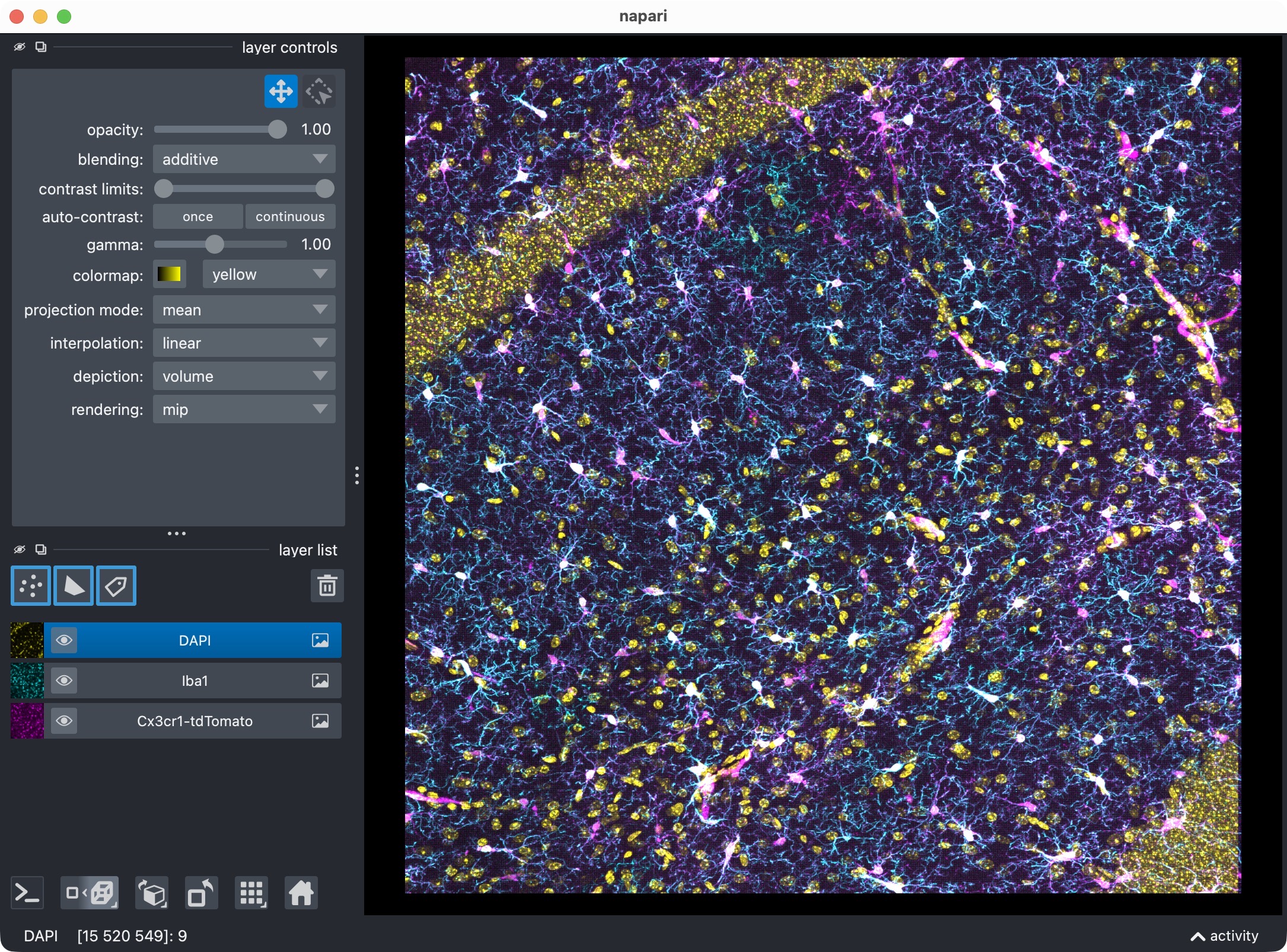
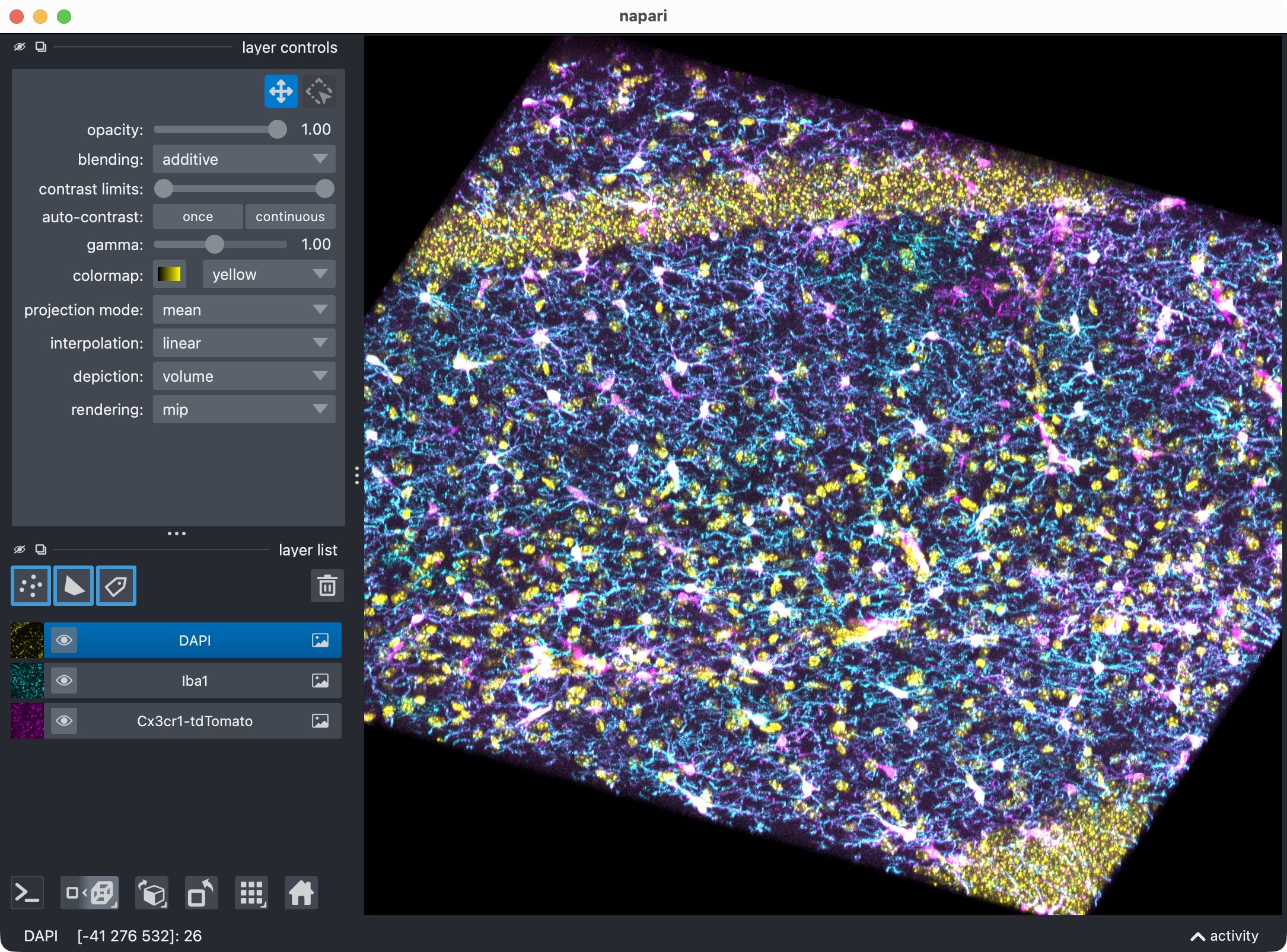
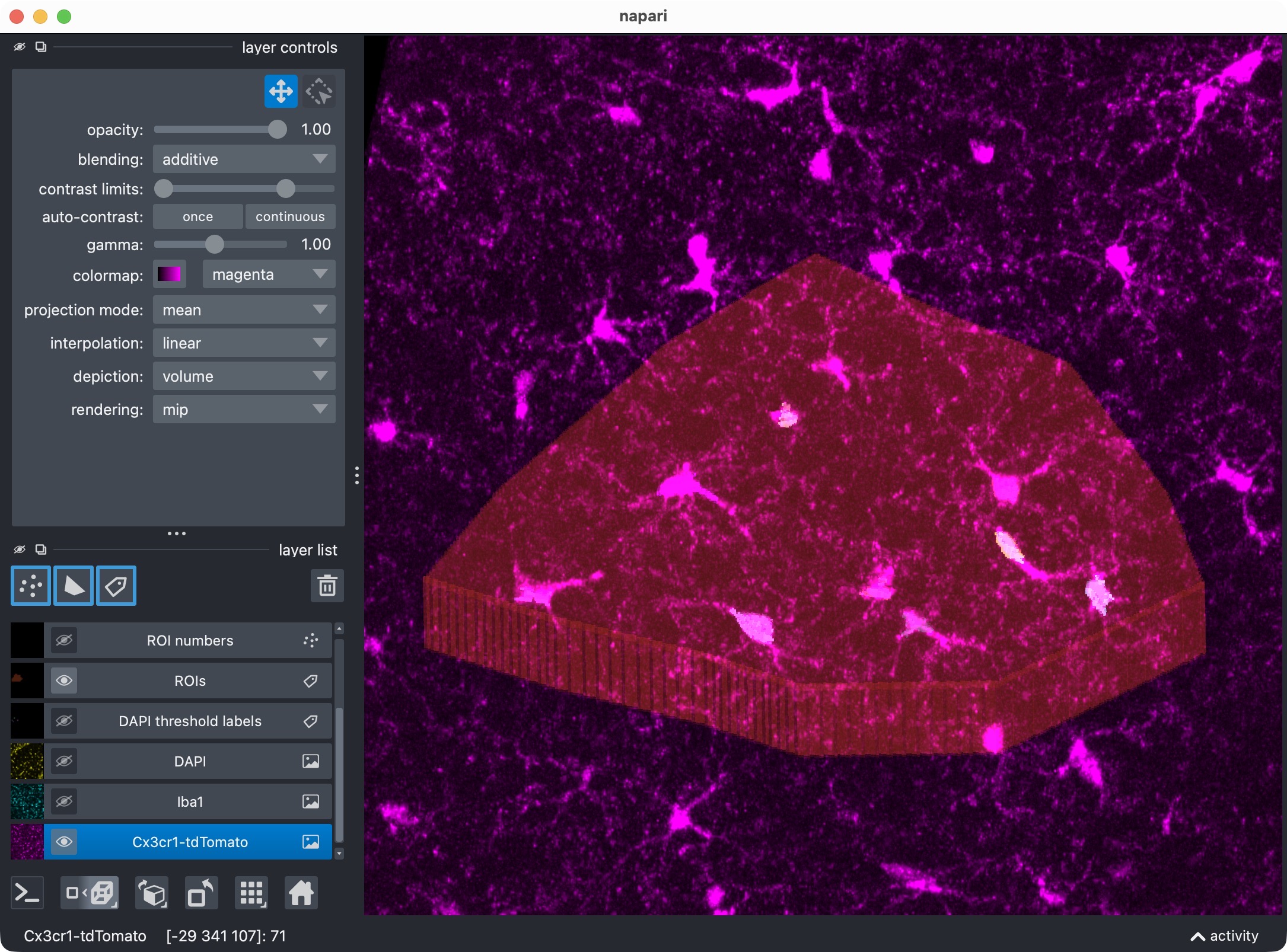
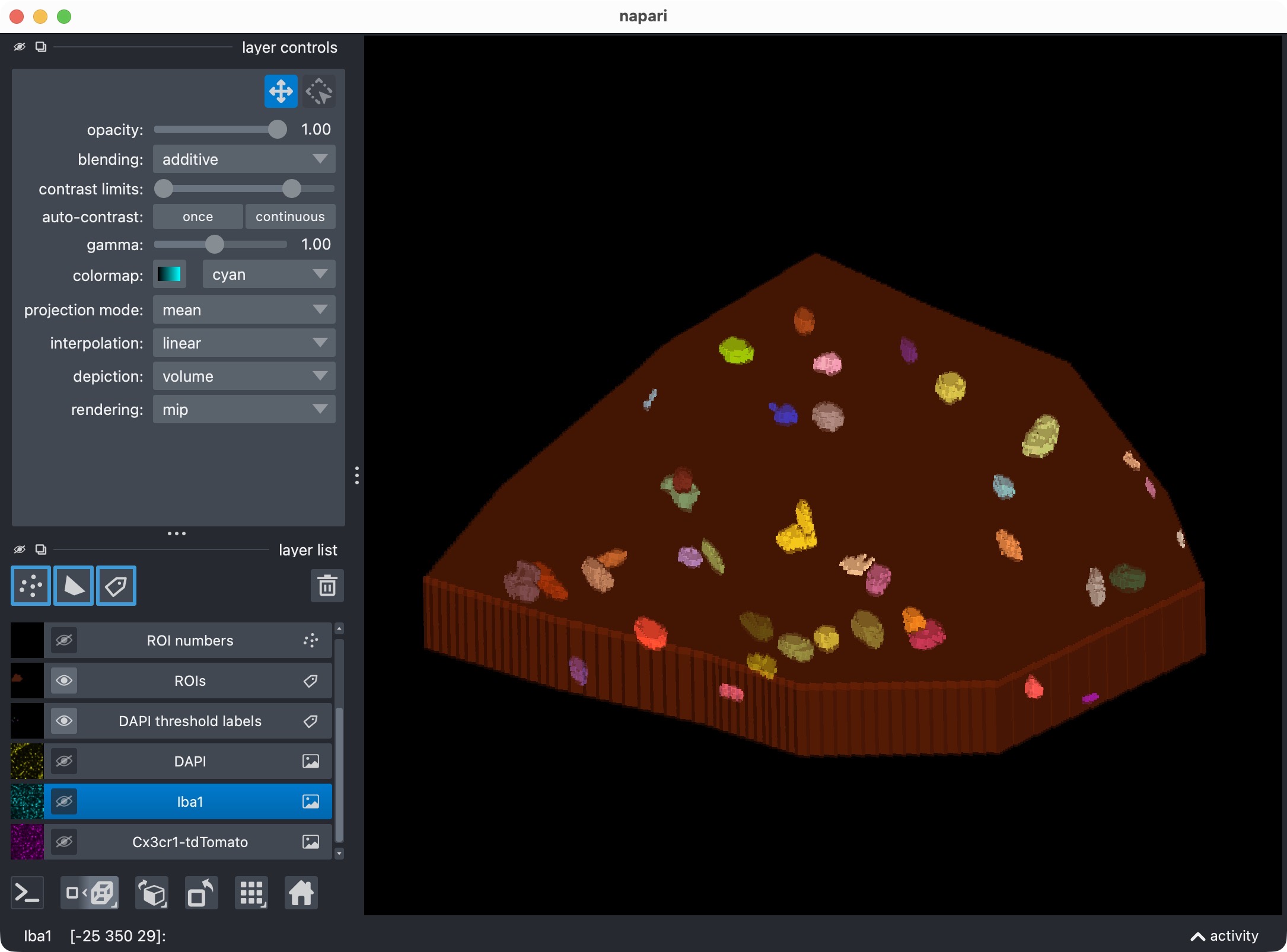
The 3D multi-channel image stack with the raw microglia (magenta), Iba1 (cyan), and DAPI (yellow) channels shown in Napari. Top shows 2D representation, bottom shows 3D representation. The DAPI channel is used for anatomical orientation but not segmented in this tutorial. The microglia channel is segmented with Cellpose, while the Iba1 channel is segmented with Otsu thresholding.
This tutorial is intentionally a feature demonstration of CellColoc’s optional third-channel analysis. It shows how a third channel can be segmented and included in the per-cell logic even if the biological meaning of that channel differs from project to project. The same structure can be reused for other 3D multichannel datasets by adapting the path selection, channel assignment, display names, and segmentation settings.
How to use this tutorial
The associated user script
user_scripts/nb_microglia_3D_three_channel_user_script.ipynb
is organized in cells, reflecting the structure of this tutorial. The same
accounts for the alternative Python script (there: # %% cells)
user_scripts/microglia_3D_three_channel_user_script.py.
The recommended way to follow this tutorial is:
open
user_scripts/nb_microglia_3D_three_channel_user_script.ipynboruser_scripts/microglia_3D_three_channel_user_script.py,run the cells from top to bottom,
adjust only the configuration values that are relevant for your own data.
The subsections below follow the same order as the script cells.
Imports
The first cell imports the public CellColoc API, napari, NumPy, and
dataclasses.replace:
from pathlib import Path
from dataclasses import replace
import napari
import numpy as np
from cellcoloc import (
CellposeModelConfig,
ChannelConfig,
ColocalizationConfig,
DisplayNames,
RuntimeConfig,
analyze_existing_masks,
build_positive_cell_mask,
create_full_image_roi_labels,
create_roi_drawing_viewer,
export_analysis_outputs,
extract_label_masks_from_viewer,
load_analysis_images,
load_roi_labels,
refine_run_result_from_cellpose_cache,
run_roi_cellpose_colocalization,
save_roi_labels_from_shapes,
show_analysis_results,
try_load_roi_labels)
PROJECT_ROOT = Path(__file__).resolve().parents[1]
What this cell does:
locates the repository root,
imports the reusable CellColoc workflow functions,
imports napari for ROI drawing and result inspection,
imports
build_positive_cell_maskto create channel-specific positivity views at the end.
Project settings
The project settings cell contains the full three-channel analysis configuration:
DATA_DIR = PROJECT_ROOT / "example_data" / "microglia_3D"
DATA_PATHS = sorted(DATA_DIR.glob("*"))
allowed_extensions = [".czi", ".tif", ".tiff", ".ome.tif", ".ome.tiff"]
DATA_PATHS = [p for p in DATA_PATHS if p.suffix.lower() in allowed_extensions]
DATA_PATHS = [p for p in DATA_PATHS if not p.name.startswith(".")]
CHANNEL_CONFIG = ChannelConfig(
cell_channel=0,
marker_channel=1,
optional_region_channel=2)
DISPLAY_NAMES = DisplayNames(
cell="Cx3cr1-tdTomato",
marker="Iba1",
optional_region="DAPI",
positive_cells="tdTomato + Iba1 positive masks")
VOXEL_SCALE_ZYX = None
CELL_MODEL_CONFIG = CellposeModelConfig(
model_name_or_path="cpsam",
segmentation_method="cellpose",
diameter=None,
z_crop=None,
anisotropy=True,
flow3d_smooth=3,
prefilter="gaussian",
prefilter_sigma_xy=0.8,
prefilter_sigma_z=0.0,
prefilter_median_size_xy=3,
prefilter_median_size_z=3,
postfilters=None,
min_intensity_measure="mean",
min_intensity_threshold=None,
local_contrast_k=1.0,
local_contrast_shell_inner_radius=1,
local_contrast_shell_outer_radius=4,
bright_pixel_measure="count",
bright_pixel_threshold=None,
bright_pixel_min_count=None,
bright_pixel_min_fraction=None,
cellprob_threshold=1.5,
flow_threshold=0.4)
MARKER_MODEL_CONFIG = CellposeModelConfig(
model_name_or_path="cpsam",
segmentation_method="cellpose",
anisotropy=True,
flow3d_smooth=3,
prefilter="laplacian_of_gaussian",
prefilter_sigma_xy=2.0,
prefilter_sigma_z=0.1,
prefilter_median_size_xy=3,
prefilter_median_size_z=3,
postfilters=None,
min_intensity_measure="mean",
min_intensity_threshold=None,
local_contrast_k=1.0,
local_contrast_shell_inner_radius=1,
local_contrast_shell_outer_radius=4,
bright_pixel_measure="count",
bright_pixel_threshold=None,
bright_pixel_min_count=None,
bright_pixel_min_fraction=None,
cellprob_threshold=0.0,
flow_threshold=0.4)
OPTIONAL_REGION_MODEL_CONFIG = CellposeModelConfig(
model_name_or_path="cpsam",
segmentation_method="cellpose",
diameter=None,
z_crop=None,
anisotropy=True,
flow3d_smooth=3,
prefilter=None,
prefilter_sigma_xy=0.0,
prefilter_sigma_z=0.0,
prefilter_median_size_xy=3,
prefilter_median_size_z=3,
postfilters=None,
min_intensity_measure="mean",
min_intensity_threshold=None,
local_contrast_k=1.0,
local_contrast_shell_inner_radius=1,
local_contrast_shell_outer_radius=4,
bright_pixel_measure="count",
bright_pixel_threshold=None,
bright_pixel_min_count=None,
bright_pixel_min_fraction=None,
cellprob_threshold=0.0,
flow_threshold=0.4)
COLOCALIZATION_CONFIG = ColocalizationConfig(
min_cell_voxels=50,
overlap_fraction_threshold=0.02,
min_overlap_voxels=10,
evaluate_optional_region_cell_positivity=True)
RUNTIME_CONFIG = RuntimeConfig(
draw_rois=True,
process_rois=True,
open_results=True,
use_gpu=True,
crop_for_testing=None,
image_loading_mode="memap")
USE_FULL_IMAGE_AS_SINGLE_ROI = False
REUSE_EXISTING_ROI_MASK_IF_AVAILABLE = True
INITIAL_RESULT_LAYER_KEYS = [
"cell_image",
"marker_image",
"optional_region_image",
"rois",
"roi_numbers",
"cell_masks",
"marker_masks",
"positive_cells",
"optional_region_labels"]
REFINEMENT_RESULT_LAYER_KEYS = [
"cell_masks",
"marker_masks",
"positive_cells",
"optional_region_labels",
"rois"]
print("Detected input files:")
for data_path in DATA_PATHS:
print(f" - {data_path.name}")
SELECTED_FILE_NAME = DATA_PATHS[0].name
DATA_PATH = DATA_DIR / SELECTED_FILE_NAME
print(f"Selected file for analysis:\n{DATA_PATH}")
This is the main cell you would adapt for your own three-channel dataset.
Input-file discovery and selection
The script scans example_data/microglia_3D/ for supported microscopy files
and lets you select one through:
SELECTED_FILE_NAME = DATA_PATHS[0].name
Channel assignment and roles
CHANNEL_CONFIG assigns three raw channels:
cell_channel=0: the channel that defines the primary segmented cell objects,marker_channel=1: the first positivity marker channel,optional_region_channel=2: the optional third analysis channel.
The naming optional_region_channel comes from the package’s generic API,
but the channel is no longer restricted to occupancy-only use cases. In this
tutorial, it is actively segmented and used in the per-cell positivity logic.
Display names
DISPLAY_NAMES controls how the channels and result layers appear in napari.
For real projects, you should replace the generic biological placeholders with
the exact marker names of your own experiment.
Voxel scale
VOXEL_SCALE_ZYX is set to None here so that CellColoc first tries to
resolve the voxel size from OMIO metadata. If needed, you can also provide it
explicitly as:
a full
(Z, Y, X)tuple for 3D datasets,or, in 2D-oriented workflows, as
(Y, X).
Three segmentation configs
This script defines three independent CellposeModelConfig objects:
CELL_MODEL_CONFIGMARKER_MODEL_CONFIGOPTIONAL_REGION_MODEL_CONFIG
In this demonstration, all three channels are segmented with Cellpose
(segmentation_method="cellpose"), even though CellColoc would also allow
threshold-based segmentation for any of them.
This is useful because it shows that the optional third channel can participate in:
segmentation,
occupancy quantification,
per-cell positivity analysis,
cache-based threshold refinement.
Third-channel cell positivity switch
The key switch for this tutorial is:
evaluate_optional_region_cell_positivity=True
inside COLOCALIZATION_CONFIG.
This tells CellColoc to do more than just quantify occupancy for the third channel. It additionally evaluates:
which channel-0 cells are positive with respect to channel 2,
and which channel-0 cells are positive for both channel 1 and channel 2.
Runtime settings and ROI mode
The runtime settings behave just like in the standard 3D tutorial:
image_loading_mode="memap"for disk-backed loading,optional ROI drawing or whole-image mode,
reuse of an existing ROI mask when available.
The script defaults to ROI-based analysis:
USE_FULL_IMAGE_AS_SINGLE_ROI = False
If you want to analyze the full field of view directly, set this to True.
Load the analysis channels
The next cell loads the selected stack and the configured channels:
loaded_images = load_analysis_images(
source_path=DATA_PATH,
channel_config=CHANNEL_CONFIG,
voxel_scale_zyx=VOXEL_SCALE_ZYX,
crop_for_testing=RUNTIME_CONFIG.crop_for_testing,
image_loading_mode=RUNTIME_CONFIG.image_loading_mode)
print(f"Results directory:\n{loaded_images.paths.results_dir}")
existing_roi_labels = None
if REUSE_EXISTING_ROI_MASK_IF_AVAILABLE:
existing_roi_labels = try_load_roi_labels(loaded_images.paths.roi_mask_path)
This step:
opens the microscopy dataset through OMIO,
extracts all three configured analysis channels,
resolves voxel size,
prepares the standardized results folder,
optionally checks whether a previously saved ROI mask already exists.
Optional ROI drawing and ROI reuse
The next two cells behave like in the other tutorials:
if USE_FULL_IMAGE_AS_SINGLE_ROI:
print("Whole-image mode is enabled. ROI drawing is skipped.")
elif existing_roi_labels is not None:
print("An existing ROI mask was found and will be reused. ROI drawing is skipped.")
elif RUNTIME_CONFIG.draw_rois:
roi_viewer, shapes_layer = create_roi_drawing_viewer(
loaded_images=loaded_images,
display_names=DISPLAY_NAMES)
print("Draw ROIs in napari and close the window. Then run the next cell.")
napari.run()
else:
print("ROI drawing is disabled. The next cell will load an existing ROI mask from disk.")
# %% SAVE THE DRAWN ROIS OR LOAD AN EXISTING ROI MASK
if USE_FULL_IMAGE_AS_SINGLE_ROI:
roi_labels_2d = create_full_image_roi_labels(loaded_images.cell_image.shape[1:])
elif existing_roi_labels is not None:
roi_labels_2d = existing_roi_labels
elif RUNTIME_CONFIG.draw_rois:
roi_labels_2d = save_roi_labels_from_shapes(
shapes_layer=shapes_layer,
output_path=loaded_images.paths.roi_mask_path,
image_shape_yx=loaded_images.cell_image.max(axis=0).shape,
scale_yx=loaded_images.voxel_scale_zyx[1:])
else:
roi_labels_2d = load_roi_labels(loaded_images.paths.roi_mask_path)
roi_ids = np.unique(roi_labels_2d)
roi_ids = roi_ids[roi_ids != 0]
print(f"ROI ids: {roi_ids}")
result_viewer = None
They support three modes:
whole-image analysis as one single ROI,
interactive ROI drawing in napari,
reuse of a previously saved ROI mask from the
results/folder.
This keeps the three-channel example fully compatible with the same interactive, reproducible ROI workflow as the other CellColoc scripts.
Run the ROI-wise three-channel segmentation and colocalization analysis
This is the main analysis step:
run_result = run_roi_cellpose_colocalization(
loaded_images=loaded_images,
roi_labels_2d=roi_labels_2d,
cell_model_config=CELL_MODEL_CONFIG,
marker_model_config=MARKER_MODEL_CONFIG,
colocalization_config=COLOCALIZATION_CONFIG,
runtime_config=RUNTIME_CONFIG,
optional_region_model_config=OPTIONAL_REGION_MODEL_CONFIG,
optional_region_result=None)
print("Initial three-channel analysis finished. "
f"Overview rows: {len(run_result.tables.overview)}, "
f"summary rows: {len(run_result.tables.summary)}.")
What happens here:
channel 0 is segmented as the primary cell-object channel,
channel 1 is segmented as the first marker channel,
channel 2 is segmented as the optional third analysis channel,
per-cell overlap is computed for channel 0 versus channel 1,
optional per-cell overlap is also computed for channel 0 versus channel 2,
ROI-level occupancy is computed for all segmented channels,
summary and overview tables are assembled.
Because evaluate_optional_region_cell_positivity=True, the summary table
contains additional columns such as:
optional_region_positivemarker_and_optional_region_positive
These are the basis for the later demonstration cells that show different positivity subsets separately.
Visualize the base result in napari
The next cell opens the base result in napari:
if RUNTIME_CONFIG.open_results:
result_viewer = show_analysis_results(
loaded_images=loaded_images,
roi_labels_2d=roi_labels_2d,
run_result=run_result,
display_names=DISPLAY_NAMES,
optional_region_result=None,
viewer=result_viewer,
layers_to_show=INITIAL_RESULT_LAYER_KEYS,
replace_existing_layers=True,
show_optional_region_image=True)
print(f"Inspecting visualization for:\n{SELECTED_FILE_NAME}")
napari.run()
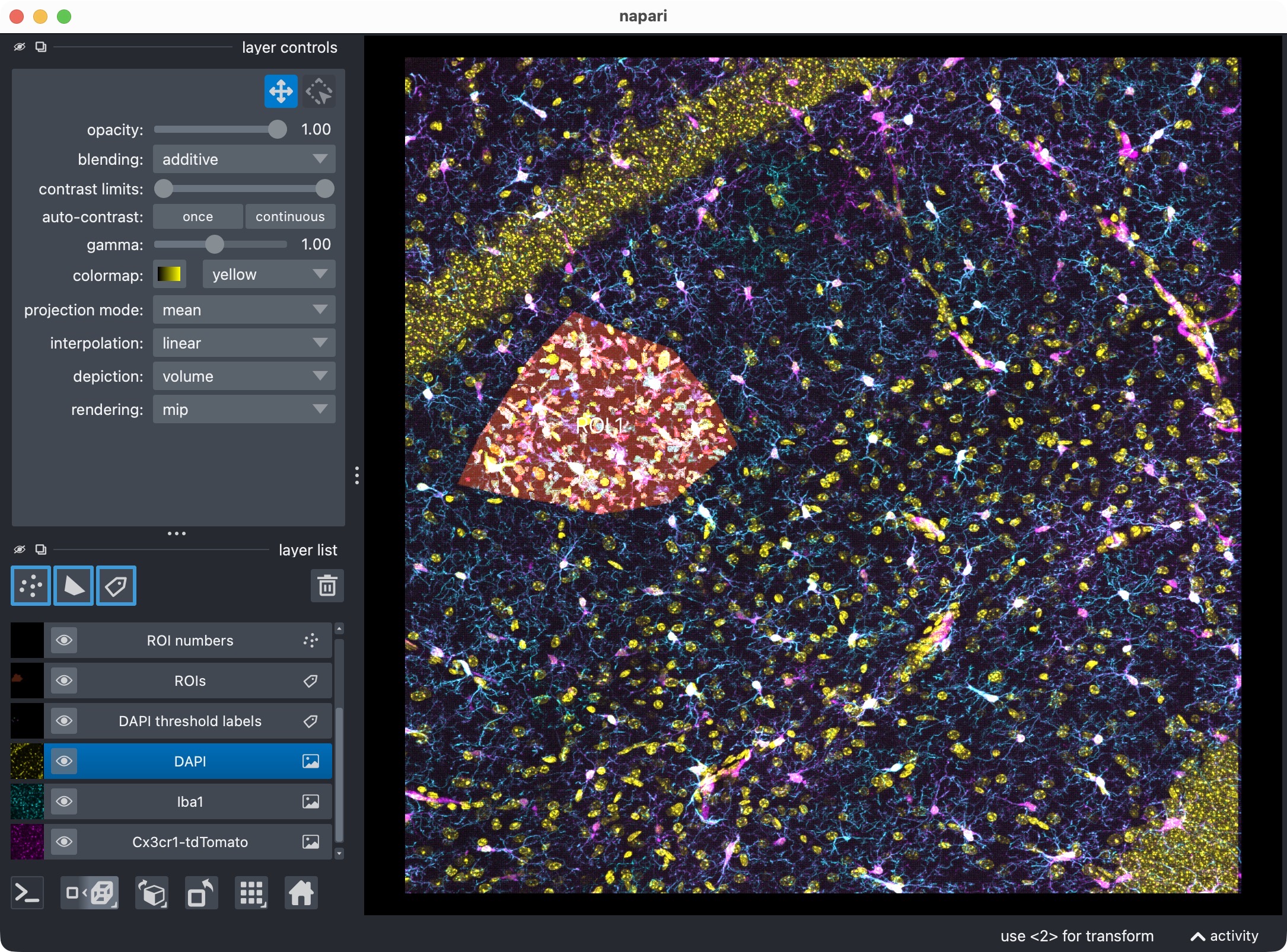
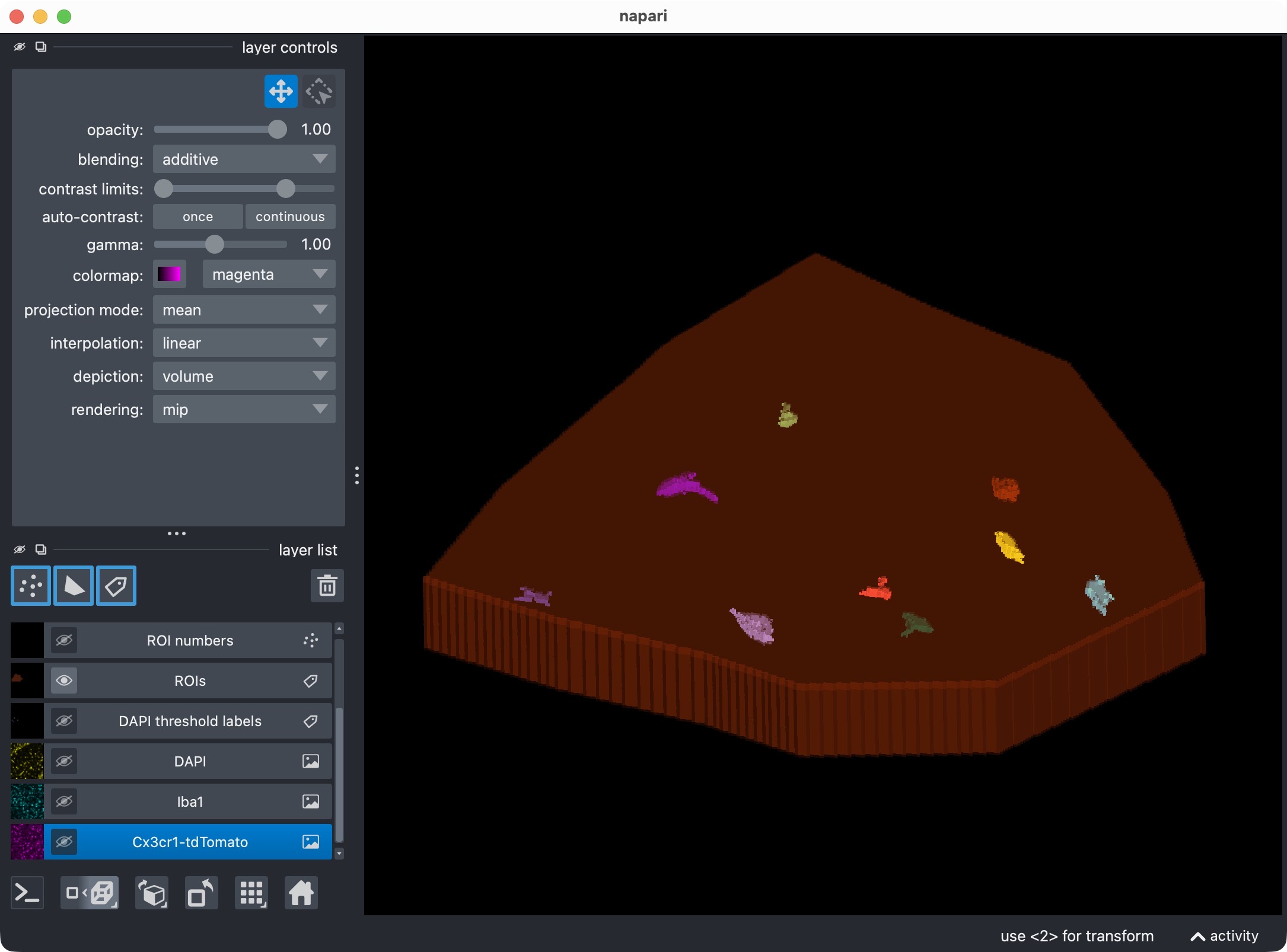
The 3D multi-channel image stack with the raw microglia (magenta), Iba1 (cyan), and DAPI (yellow) channels shown in Napari. In this tutorial, we also segmented the optional third channel (DAPI) and included it in the per-cell positivity analysis. The microglia channel is segmented with Cellpose, while the Iba1 channel is segmented with Otsu thresholding.
This viewer can show:
the raw cell channel,
the raw marker channel,
the raw third channel,
ROI labels,
cell masks,
marker masks,
positive-cell masks,
the segmented third-channel labels.
This is the main checkpoint where you can verify that all three segmentation paths look plausible before moving on to refinement.
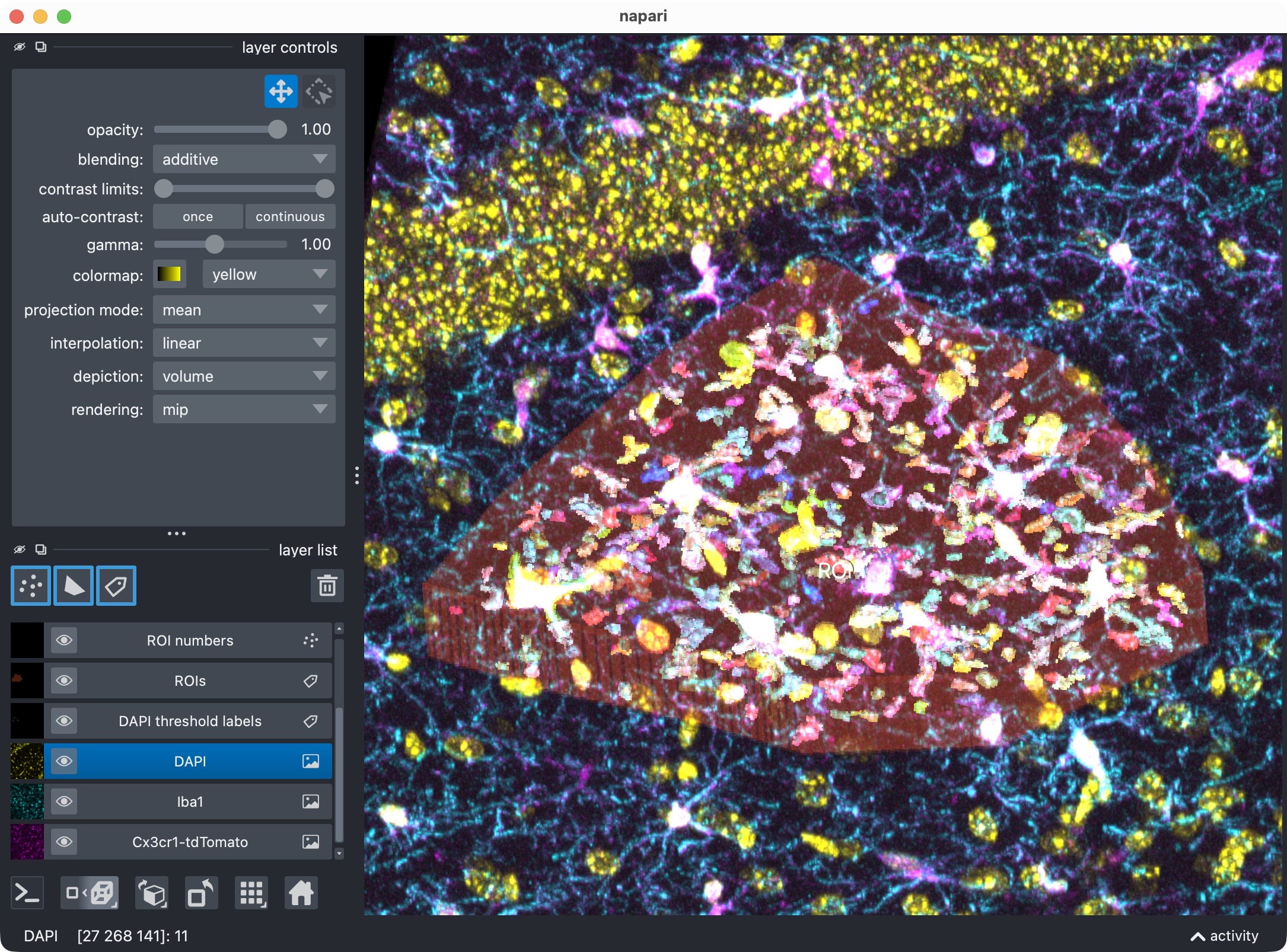
Top: Zoom onto the analyzed ROI, showing all channels and their segmented label layers. Center: Microglia channel only. Bottom: Segmentation layer of the microglia channel, showing the Cellpose-segmented cell objects. Note that we miss the upper center microglia cell, while the remaining cells are segmented correctly. This is a typical Cellpose segmentation result that can be improved with the optional refinement step.
Top: Zoom onto the analyzed ROI, showing the marker channel and its segmented label layer. Bottom: Marker channel only. The marker channel is segmented with Otsu thresholding, which results in a rather rough mask. However, since we are only interested in per-cell positivity (which microglia is Iba1-positive?), this is sufficient for the current demonstration.
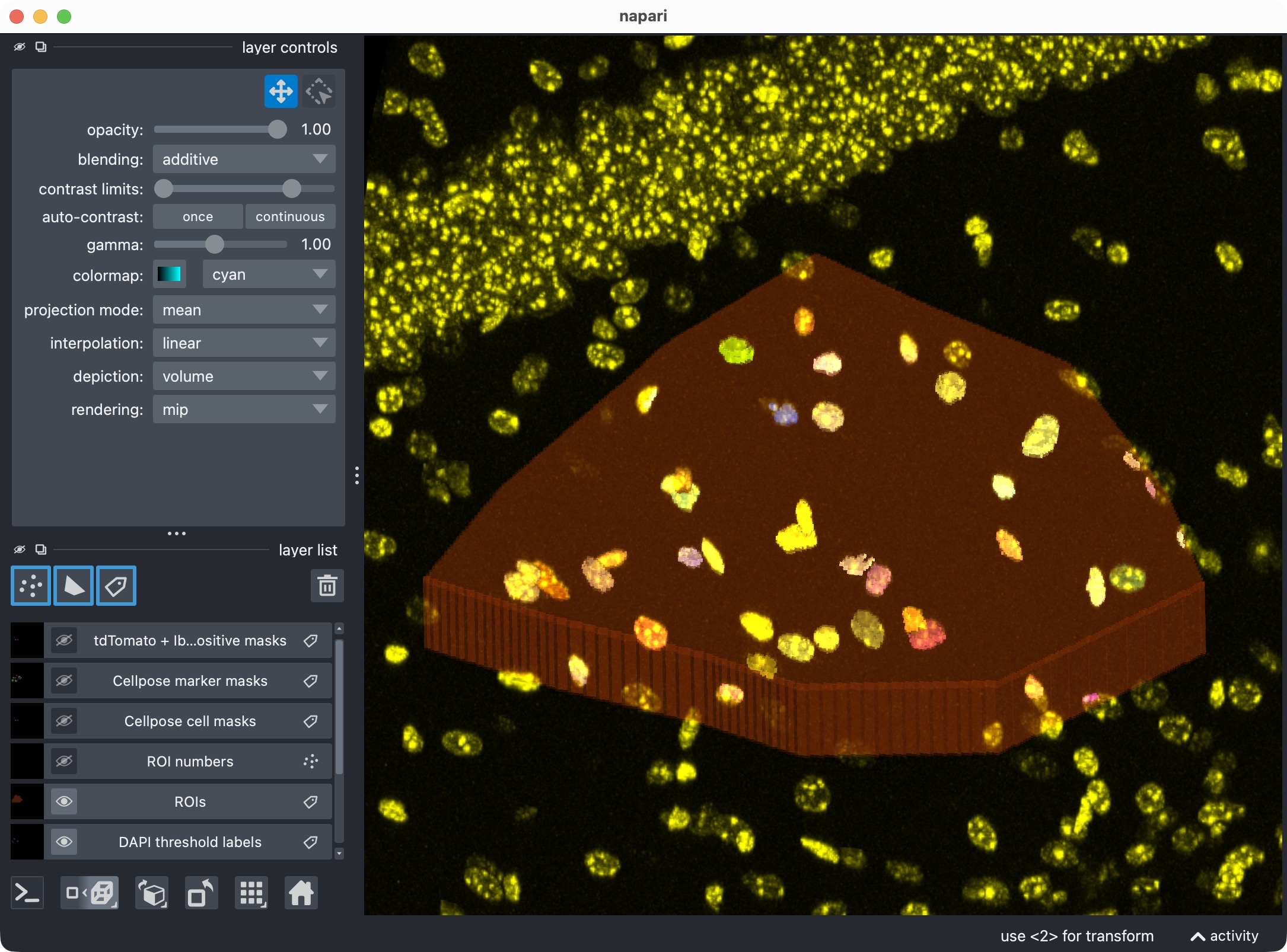
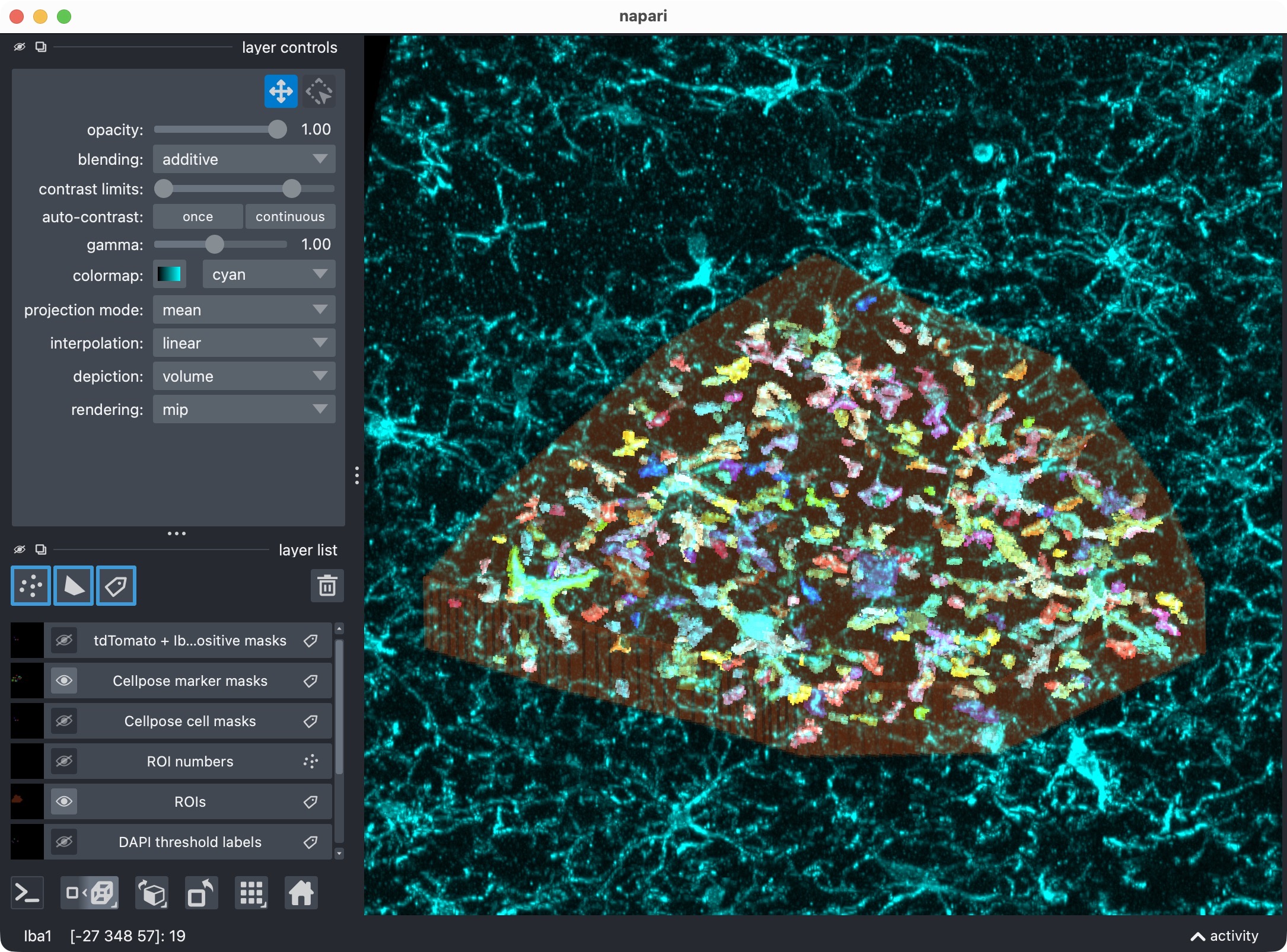
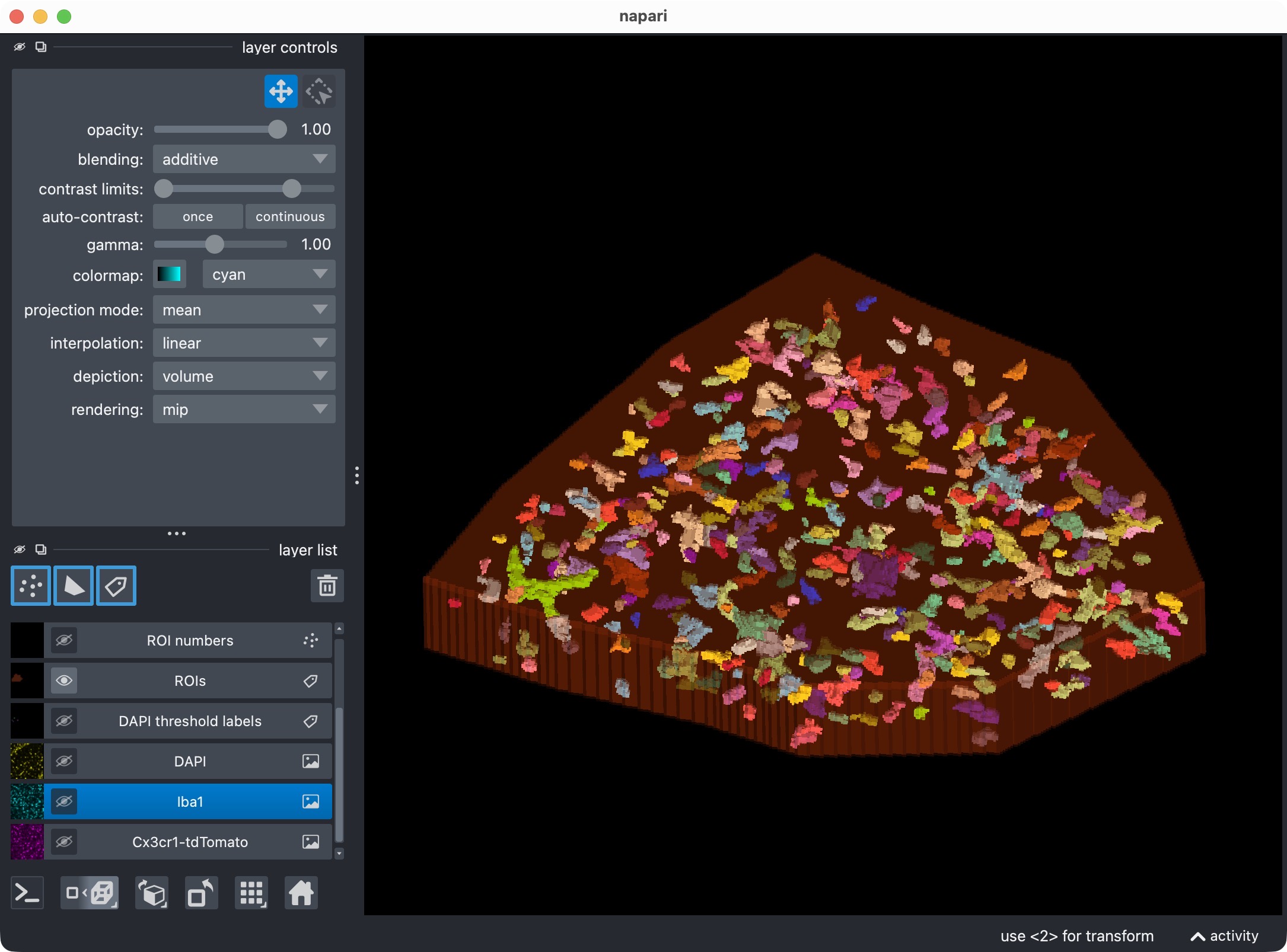
Top: Zoom onto the analyzed ROI, showing the optional third channel and its segmented label layer. Bottom: Segmented third channel only. The optional third channel is segmented with Cellpose, which results in an almost perfect mask this time. This shows that the optional third channel can be segmented and included in the per-cell positivity analysis, even if its biological meaning differs from the first marker channel. It also demonstrates that Cellpose’s segmentation quality can vary across channels and tends to work best for more “roundish” objects (the microglia’s processes complicate the Cellpose segmentation).
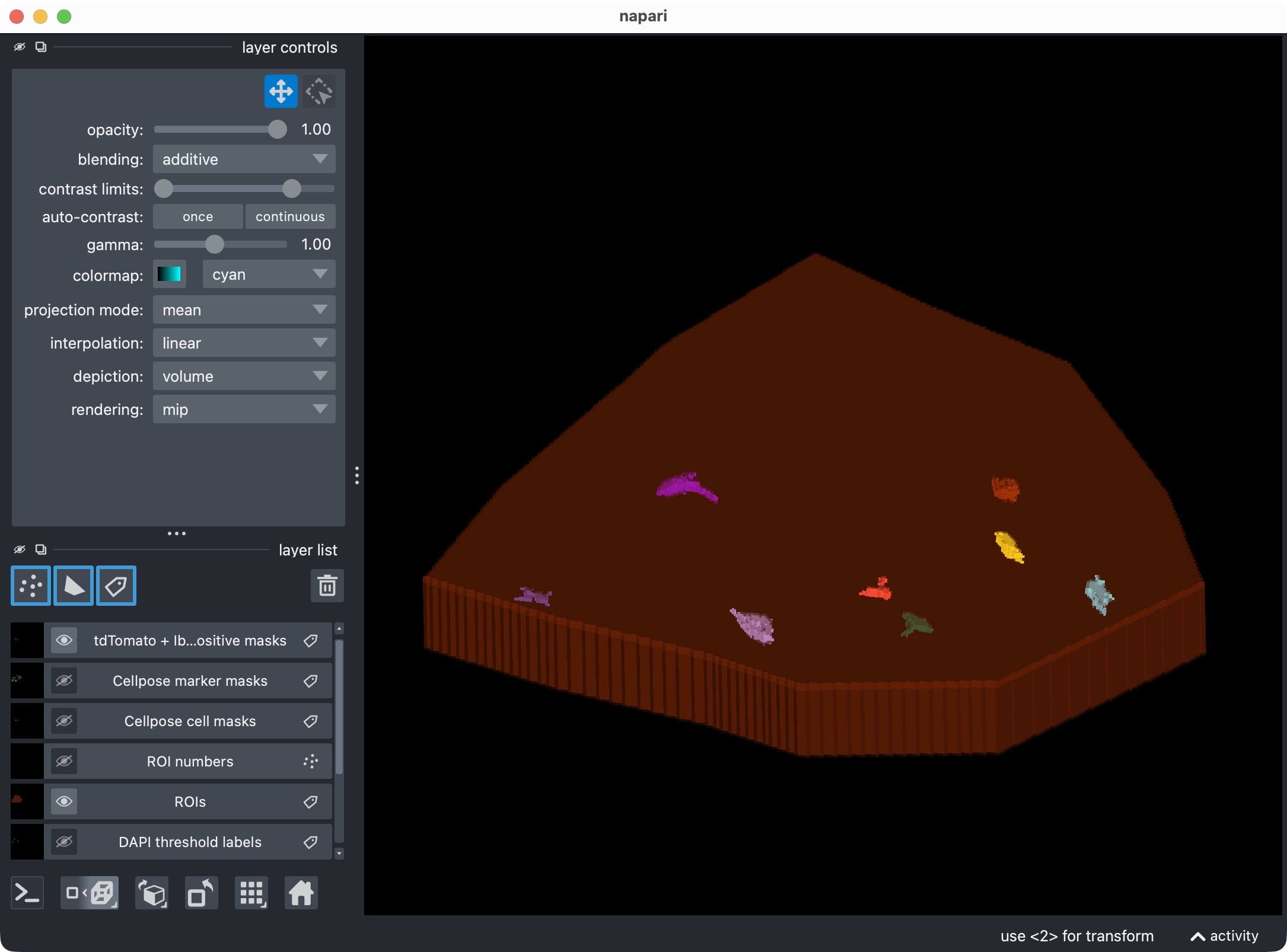
Microglia segmentation layer, including only cells that are both Iba1-positive and DAPI-positive. This is the most specific view of the three-channel analysis, showing only microglia that are positive for both marker channels. It demonstrates that the optional third channel can be used to refine the per-cell positivity analysis and create more specific subsets of cells.
Optional global z-crop for refinement
The next cell lets you define or update a global z interval for the following refinement step:
REFINEMENT_ANALYSIS_Z_CROP = None
if REFINEMENT_ANALYSIS_Z_CROP is None:
print("No refinement z crop requested. Subsequent refinement will keep the "
f"current analysis z range: {run_result.analysis_z_bounds}.")
else:
print("Subsequent refinement will use this global analysis z crop:\n"
f"{REFINEMENT_ANALYSIS_Z_CROP}")
As in the regular 3D tutorial, this allows you to:
inspect the full 3D result first,
then restrict refinement and quantification to a chosen z interval,
while keeping the original XY ROI definition unchanged.
Optionally refine all three channels and visualize the updated result
The next cell demonstrates cache-based threshold refinement for all three Cellpose-segmented channels:
REFINE_WITH_CACHED_CELLPOSE_OUTPUTS = False
REFINED_CELL_CELLPROB_THRESHOLD = CELL_MODEL_CONFIG.cellprob_threshold - 3.0
REFINED_CELL_FLOW_THRESHOLD = CELL_MODEL_CONFIG.flow_threshold - 0.2
REFINED_MARKER_CELLPROB_THRESHOLD = MARKER_MODEL_CONFIG.cellprob_threshold - 3.0
REFINED_MARKER_FLOW_THRESHOLD = MARKER_MODEL_CONFIG.flow_threshold
REFINED_OPTIONAL_REGION_CELLPROB_THRESHOLD = (OPTIONAL_REGION_MODEL_CONFIG.cellprob_threshold - 3.0)
REFINED_OPTIONAL_REGION_FLOW_THRESHOLD = OPTIONAL_REGION_MODEL_CONFIG.flow_threshold
REFINED_CELL_POSTFILTERS = ["min_intensity", "bright_pixel_support", "local_contrast"]
REFINED_CELL_MIN_INTENSITY_MEASURE = "max"
REFINED_CELL_MIN_INTENSITY_THRESHOLD = 250
REFINED_CELL_LOCAL_CONTRAST_K = 4
REFINED_CELL_LOCAL_CONTRAST_SHELL_INNER_RADIUS = 1.0
REFINED_CELL_LOCAL_CONTRAST_SHELL_OUTER_RADIUS = 10
REFINED_CELL_BRIGHT_PIXEL_MEASURE = "fraction"
REFINED_CELL_BRIGHT_PIXEL_THRESHOLD = 110
REFINED_CELL_BRIGHT_PIXEL_MIN_COUNT = None
REFINED_CELL_BRIGHT_PIXEL_MIN_FRACTION = 0.08
REFINED_MARKER_POSTFILTERS = ["min_intensity", "bright_pixel_support", "local_contrast"]
REFINED_MARKER_MIN_INTENSITY_MEASURE = "max"
REFINED_MARKER_MIN_INTENSITY_THRESHOLD = 250
REFINED_MARKER_LOCAL_CONTRAST_K = 1
REFINED_MARKER_LOCAL_CONTRAST_SHELL_INNER_RADIUS = 1.0
REFINED_MARKER_LOCAL_CONTRAST_SHELL_OUTER_RADIUS = 4
REFINED_MARKER_BRIGHT_PIXEL_MEASURE = "fraction"
REFINED_MARKER_BRIGHT_PIXEL_THRESHOLD = 120
REFINED_MARKER_BRIGHT_PIXEL_MIN_COUNT = None
REFINED_MARKER_BRIGHT_PIXEL_MIN_FRACTION = 0.09
REFINED_OPTIONAL_REGION_POSTFILTERS = ["min_intensity", "bright_pixel_support", "local_contrast"]
REFINED_OPTIONAL_REGION_MIN_INTENSITY_MEASURE = "max"
REFINED_OPTIONAL_REGION_MIN_INTENSITY_THRESHOLD = 250
REFINED_OPTIONAL_REGION_LOCAL_CONTRAST_K = 1
REFINED_OPTIONAL_REGION_LOCAL_CONTRAST_SHELL_INNER_RADIUS = 1.0
REFINED_OPTIONAL_REGION_LOCAL_CONTRAST_SHELL_OUTER_RADIUS = 4
REFINED_OPTIONAL_REGION_BRIGHT_PIXEL_MEASURE = "fraction"
REFINED_OPTIONAL_REGION_BRIGHT_PIXEL_THRESHOLD = 120
REFINED_OPTIONAL_REGION_BRIGHT_PIXEL_MIN_COUNT = None
REFINED_OPTIONAL_REGION_BRIGHT_PIXEL_MIN_FRACTION = 0.09
effective_refinement_z_crop = (
CELL_MODEL_CONFIG.z_crop if REFINEMENT_ANALYSIS_Z_CROP is None else REFINEMENT_ANALYSIS_Z_CROP)
if REFINE_WITH_CACHED_CELLPOSE_OUTPUTS:
refined_cell_model_config = replace(
CELL_MODEL_CONFIG,
z_crop=effective_refinement_z_crop,
postfilters=REFINED_CELL_POSTFILTERS,
min_intensity_measure=REFINED_CELL_MIN_INTENSITY_MEASURE,
min_intensity_threshold=REFINED_CELL_MIN_INTENSITY_THRESHOLD,
local_contrast_k=REFINED_CELL_LOCAL_CONTRAST_K,
local_contrast_shell_inner_radius=REFINED_CELL_LOCAL_CONTRAST_SHELL_INNER_RADIUS,
local_contrast_shell_outer_radius=REFINED_CELL_LOCAL_CONTRAST_SHELL_OUTER_RADIUS,
bright_pixel_measure=REFINED_CELL_BRIGHT_PIXEL_MEASURE,
bright_pixel_threshold=REFINED_CELL_BRIGHT_PIXEL_THRESHOLD,
bright_pixel_min_count=REFINED_CELL_BRIGHT_PIXEL_MIN_COUNT,
bright_pixel_min_fraction=REFINED_CELL_BRIGHT_PIXEL_MIN_FRACTION)
refined_marker_model_config = replace(
MARKER_MODEL_CONFIG,
z_crop=effective_refinement_z_crop,
postfilters=REFINED_MARKER_POSTFILTERS,
min_intensity_measure=REFINED_MARKER_MIN_INTENSITY_MEASURE,
min_intensity_threshold=REFINED_MARKER_MIN_INTENSITY_THRESHOLD,
local_contrast_k=REFINED_MARKER_LOCAL_CONTRAST_K,
local_contrast_shell_inner_radius=REFINED_MARKER_LOCAL_CONTRAST_SHELL_INNER_RADIUS,
local_contrast_shell_outer_radius=REFINED_MARKER_LOCAL_CONTRAST_SHELL_OUTER_RADIUS,
bright_pixel_measure=REFINED_MARKER_BRIGHT_PIXEL_MEASURE,
bright_pixel_threshold=REFINED_MARKER_BRIGHT_PIXEL_THRESHOLD,
bright_pixel_min_count=REFINED_MARKER_BRIGHT_PIXEL_MIN_COUNT,
bright_pixel_min_fraction=REFINED_MARKER_BRIGHT_PIXEL_MIN_FRACTION)
refined_optional_region_model_config = replace(
OPTIONAL_REGION_MODEL_CONFIG,
z_crop=effective_refinement_z_crop,
postfilters=REFINED_OPTIONAL_REGION_POSTFILTERS,
min_intensity_measure=REFINED_OPTIONAL_REGION_MIN_INTENSITY_MEASURE,
min_intensity_threshold=REFINED_OPTIONAL_REGION_MIN_INTENSITY_THRESHOLD,
local_contrast_k=REFINED_OPTIONAL_REGION_LOCAL_CONTRAST_K,
local_contrast_shell_inner_radius=REFINED_OPTIONAL_REGION_LOCAL_CONTRAST_SHELL_INNER_RADIUS,
local_contrast_shell_outer_radius=REFINED_OPTIONAL_REGION_LOCAL_CONTRAST_SHELL_OUTER_RADIUS,
bright_pixel_measure=REFINED_OPTIONAL_REGION_BRIGHT_PIXEL_MEASURE,
bright_pixel_threshold=REFINED_OPTIONAL_REGION_BRIGHT_PIXEL_THRESHOLD,
bright_pixel_min_count=REFINED_OPTIONAL_REGION_BRIGHT_PIXEL_MIN_COUNT,
bright_pixel_min_fraction=REFINED_OPTIONAL_REGION_BRIGHT_PIXEL_MIN_FRACTION)
run_result = refine_run_result_from_cellpose_cache(
loaded_images=loaded_images,
roi_labels_2d=roi_labels_2d,
run_result=run_result,
colocalization_config=COLOCALIZATION_CONFIG,
cell_model_config=refined_cell_model_config,
marker_model_config=refined_marker_model_config,
optional_region_model_config=refined_optional_region_model_config,
cell_cellprob_threshold=REFINED_CELL_CELLPROB_THRESHOLD,
cell_flow_threshold=REFINED_CELL_FLOW_THRESHOLD,
marker_cellprob_threshold=REFINED_MARKER_CELLPROB_THRESHOLD,
marker_flow_threshold=REFINED_MARKER_FLOW_THRESHOLD,
optional_region_cellprob_threshold=REFINED_OPTIONAL_REGION_CELLPROB_THRESHOLD,
optional_region_flow_threshold=REFINED_OPTIONAL_REGION_FLOW_THRESHOLD,
optional_region_result=None)
print("Refinement finished. "
f"Overview rows: {len(run_result.tables.overview)}, "
f"summary rows: {len(run_result.tables.summary)}.")
if RUNTIME_CONFIG.open_results:
result_viewer = show_analysis_results(
loaded_images=loaded_images,
roi_labels_2d=roi_labels_2d,
run_result=run_result,
display_names=DISPLAY_NAMES,
optional_region_result=None,
viewer=result_viewer,
layers_to_show=REFINEMENT_RESULT_LAYER_KEYS,
replace_existing_layers=True,
show_optional_region_image=True)
print(f"Inspecting refined visualization for:\n{SELECTED_FILE_NAME}")
napari.run()
else:
print("Cached Cellpose refinement is disabled for this run.")
Thus, the optional third analysis channel can be refined in the same way as the first two channels. Overall, the cell demonstrates:
Cellpose threshold refinement for the cell channel,
Cellpose threshold refinement for the marker channel,
Cellpose threshold refinement for the third channel,
optional postfilters for all three channels,
optional refinement-time z cropping,
viewer refresh with the updated three-channel result.
Important refinement parameters are:
REFINED_CELL_*REFINED_MARKER_*REFINED_OPTIONAL_REGION_*
Each group contains:
CELLPROB_THRESHOLDFLOW_THRESHOLDoptional postfilter selector
optional postfilter parameters
This makes the three-channel script a good template for experiments in which all analyzed channels should remain editable and tunable after the initial segmentation run. For a detailled walkthrough through each refinement step, please refer to the 3D data analysis tutorial.
Optional manual reanalysis after napari edits
The next cell supports manual label editing followed by full reanalysis:
REANALYZE_EDITED_LABELS_FROM_VIEWER = False
if REANALYZE_EDITED_LABELS_FROM_VIEWER:
cell_masks_from_viewer, marker_masks_from_viewer = extract_label_masks_from_viewer(result_viewer)
run_result = analyze_existing_masks(
loaded_images=loaded_images,
roi_labels_2d=roi_labels_2d,
cell_masks=cell_masks_from_viewer,
marker_masks=marker_masks_from_viewer,
colocalization_config=COLOCALIZATION_CONFIG,
optional_region_result=None,
optional_region_masks=run_result.optional_region_masks,
analysis_z_bounds=run_result.analysis_z_bounds,
cell_refinement_context=run_result.cell_refinement_context,
marker_refinement_context=run_result.marker_refinement_context)
result_viewer = show_analysis_results(
loaded_images=loaded_images,
roi_labels_2d=roi_labels_2d,
run_result=run_result,
display_names=DISPLAY_NAMES,
optional_region_result=None,
viewer=result_viewer,
layers_to_show=REFINEMENT_RESULT_LAYER_KEYS,
replace_existing_layers=True,
show_optional_region_image=True,)
print("Inspect the relabeled result in napari and close the window when finished.")
napari.run()
else:
print("Manual label reanalysis from the napari viewer is disabled for this run.")
This behaves similarly to the manual reanalysis in the standard 3D tutorial.
In the current demo, the manual reanalysis can read back the edited cell and
marker layers. The third-channel masks are reused from the current
run_result. For a detailled walkthrough, please
refer to the 3D data analysis tutorial.
Visualize cells positive for channel 0 + channel 1
The next cell creates a positivity mask for cells that are positive with respect to channel 1:
summary_channel01 = run_result.tables.summary.copy()
summary_channel01["marker_positive"] = summary_channel01["marker_positive"].astype(bool)
channel01_positive_masks = build_positive_cell_mask(run_result.cell_masks, summary_channel01)
viewer_01 = napari.Viewer()
viewer_01.add_image(
loaded_images.cell_image,
name=DISPLAY_NAMES.cell,
scale=loaded_images.voxel_scale_zyx,
blending="additive",
colormap="magenta")
viewer_01.add_image(
loaded_images.marker_image,
name=DISPLAY_NAMES.marker,
scale=loaded_images.voxel_scale_zyx,
blending="additive",
colormap="green")
viewer_01.add_labels(
channel01_positive_masks,
name="Cells positive for channel 0 + channel 1",
scale=loaded_images.voxel_scale_zyx,
blending="additive")
print("Inspect channel-0 plus channel-1 positive cells.")
napari.run()
Internally, this uses:
run_result.tables.summary["marker_positive"]and
build_positive_cell_mask(...)
This is the standard two-channel positivity view which you get when analyzing only two channels.
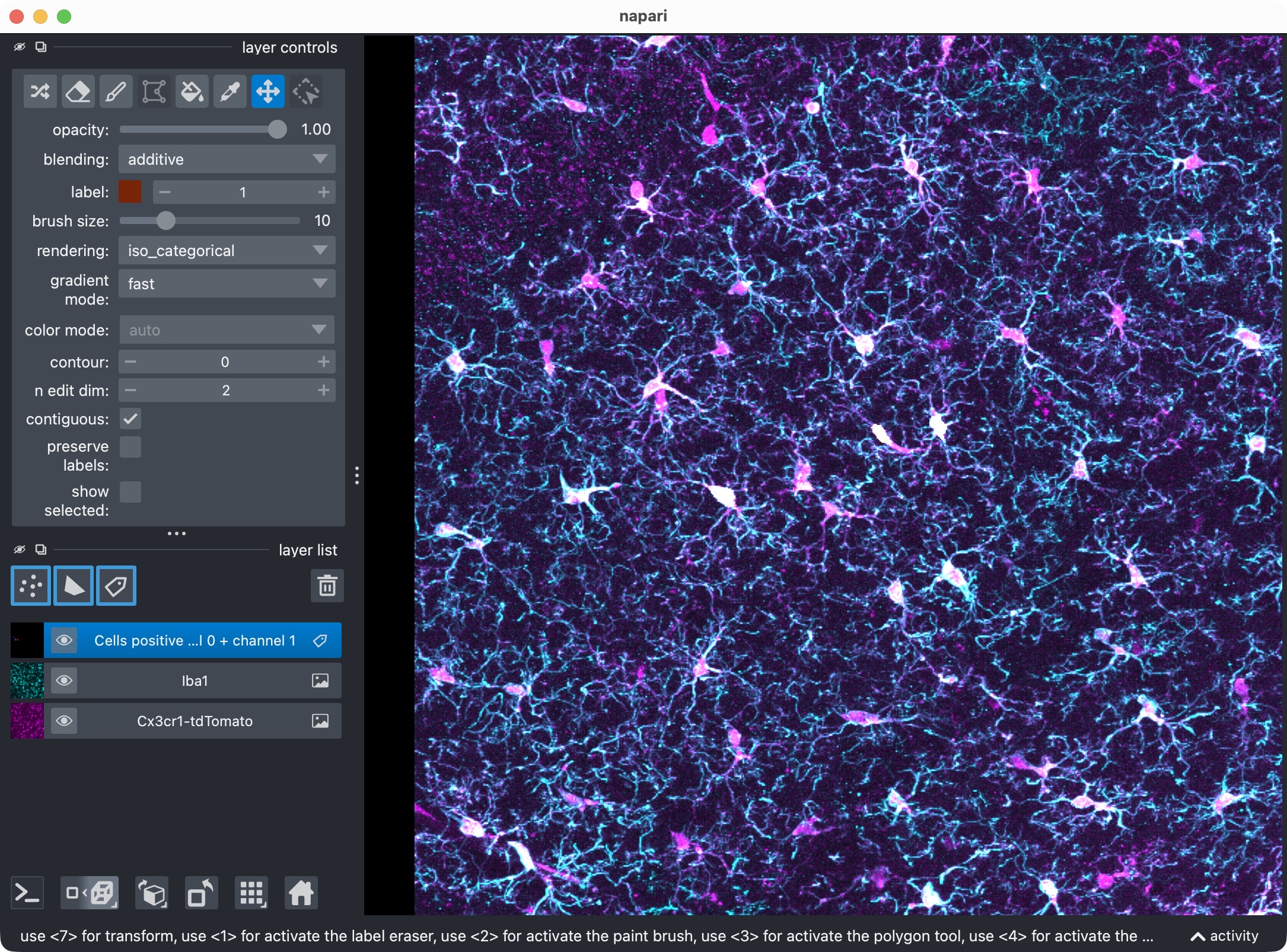
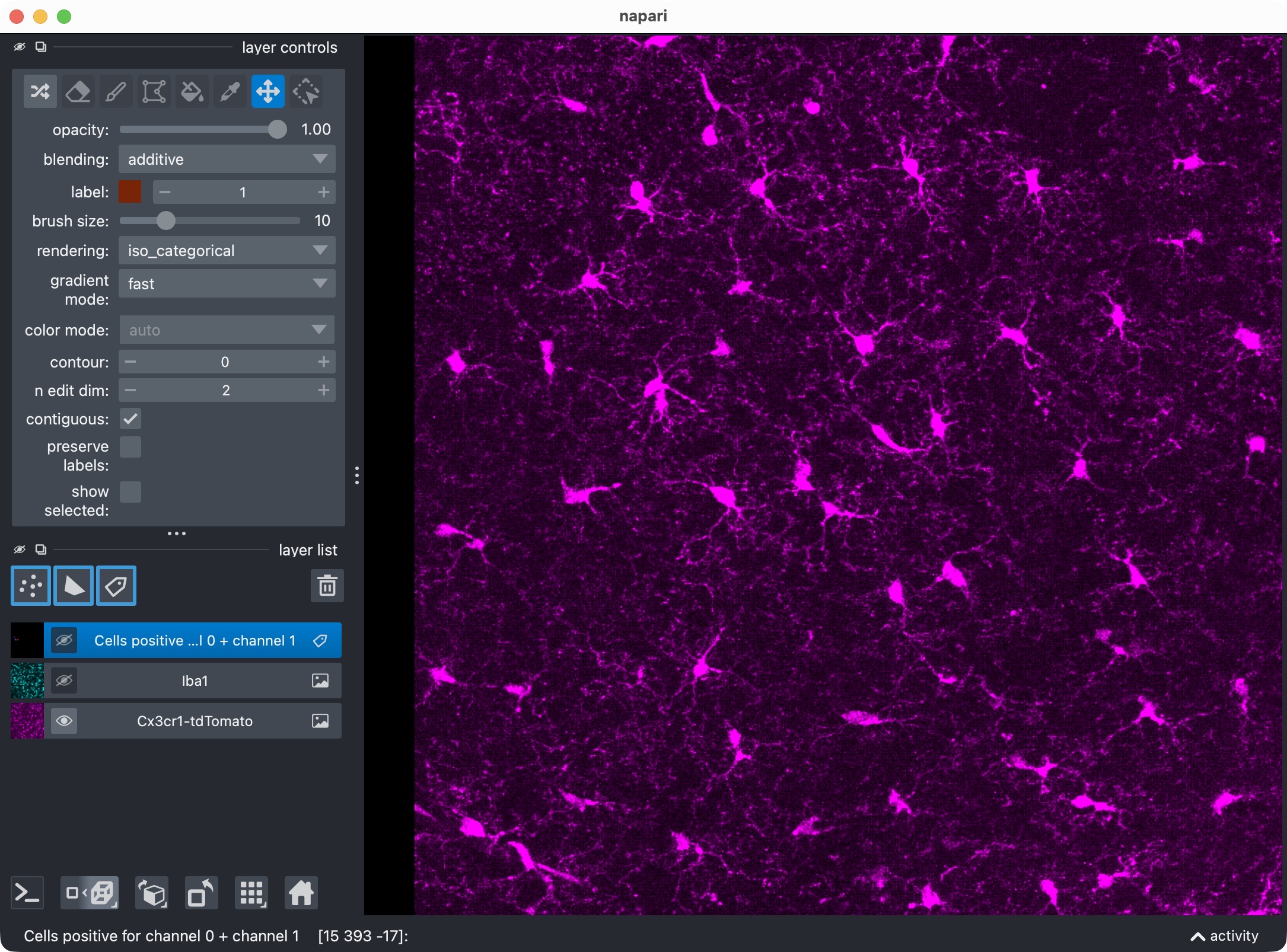
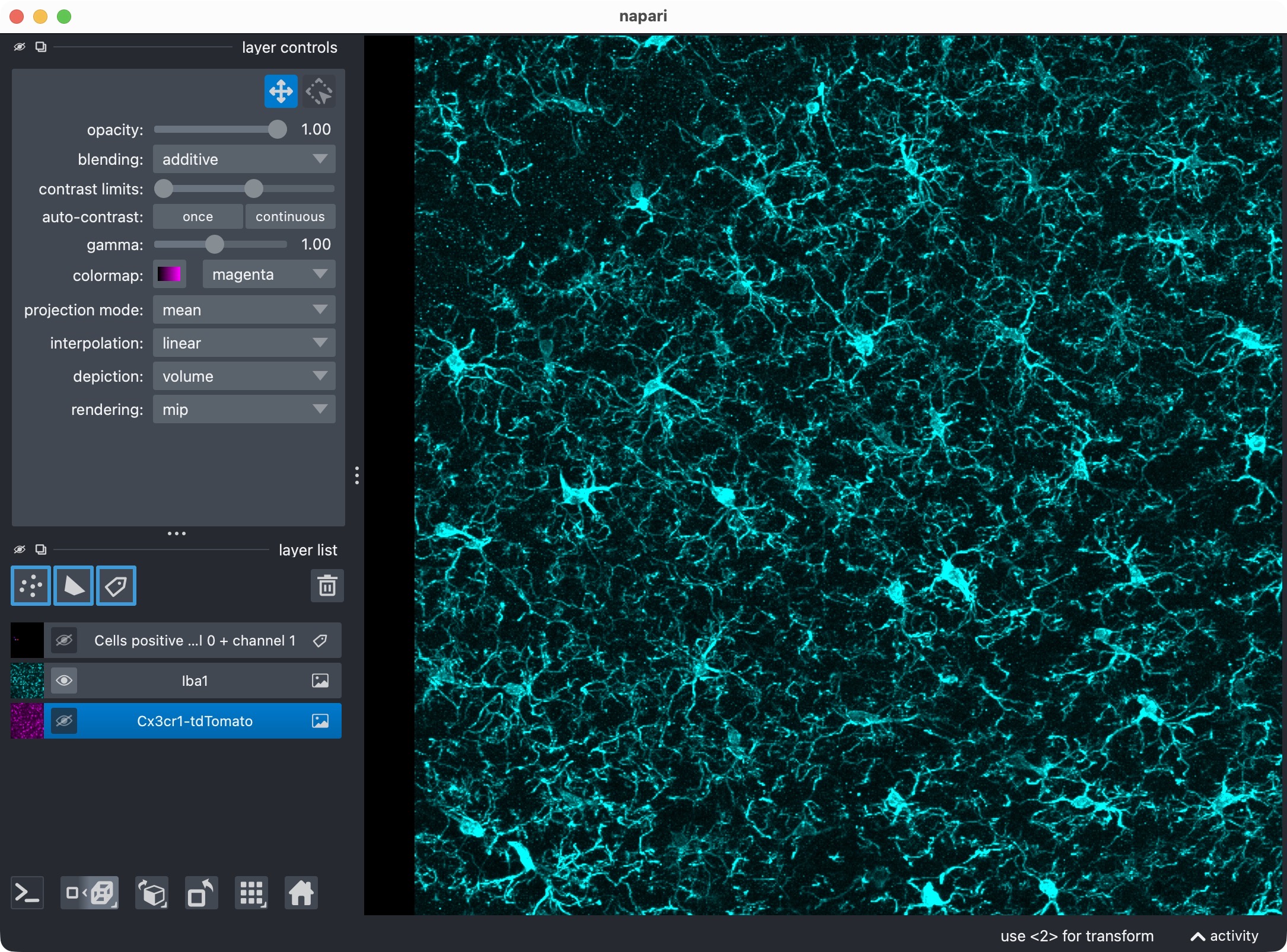
Top: Overview of the microglia (magenta) and Iba1 (cyan) channels. Center top: The microglia channel only. Center bottom: The Iba1 channel only. Bottom: Segmentation layer of the Iba1-positive cells.
Visualize cells positive for channel 0 + channel 2
The next cell creates a positivity mask for cells that are positive with respect to the optional third channel:
summary_channel02 = run_result.tables.summary.copy()
summary_channel02["marker_positive"] = summary_channel02["optional_region_positive"].astype(bool)
channel02_positive_masks = build_positive_cell_mask(run_result.cell_masks, summary_channel02)
viewer_02 = napari.Viewer()
viewer_02.add_image(
loaded_images.cell_image,
name=DISPLAY_NAMES.cell,
scale=loaded_images.voxel_scale_zyx,
blending="additive",
colormap="magenta")
viewer_02.add_image(
loaded_images.optional_region_image,
name=DISPLAY_NAMES.optional_region,
scale=loaded_images.voxel_scale_zyx,
blending="additive",
colormap="yellow")
viewer_02.add_labels(
channel02_positive_masks,
name="Cells positive for channel 0 + channel 2",
scale=loaded_images.voxel_scale_zyx,
blending="additive")
print("Inspect channel-0 plus channel-2 positive cells.")
napari.run()
Here the script temporarily maps:
summary["optional_region_positive"]
onto the marker_positive column expected by
build_positive_cell_mask(...).
This gives you a clean standalone view of channel-0 cells that are positive for the third channel, independently of channel 1.
Visualize cells positive for channel 0 + channel 1 + channel 2
The final dedicated positivity-view cell shows cells that are positive for both marker channels:
summary_channel012 = run_result.tables.summary.copy()
summary_channel012["marker_positive"] = summary_channel012["marker_and_optional_region_positive"].astype(bool)
channel012_positive_masks = build_positive_cell_mask(run_result.cell_masks, summary_channel012)
viewer_012 = napari.Viewer()
viewer_012.add_image(
loaded_images.cell_image,
name=DISPLAY_NAMES.cell,
scale=loaded_images.voxel_scale_zyx,
blending="additive",
colormap="magenta")
viewer_012.add_image(
loaded_images.marker_image,
name=DISPLAY_NAMES.marker,
scale=loaded_images.voxel_scale_zyx,
blending="additive",
colormap="green")
viewer_012.add_image(
loaded_images.optional_region_image,
name=DISPLAY_NAMES.optional_region,
scale=loaded_images.voxel_scale_zyx,
blending="additive",
colormap="yellow")
viewer_012.add_labels(
channel012_positive_masks,
name="Cells positive for channel 0 + channel 1 + channel 2",
scale=loaded_images.voxel_scale_zyx,
blending="additive")
print("Inspect channel-0 plus channel-1 plus channel-2 positive cells.")
napari.run()
This uses:
summary["marker_and_optional_region_positive"]
and therefore corresponds to the logical AND of:
channel-0 versus channel-1 positivity,
channel-0 versus channel-2 positivity.
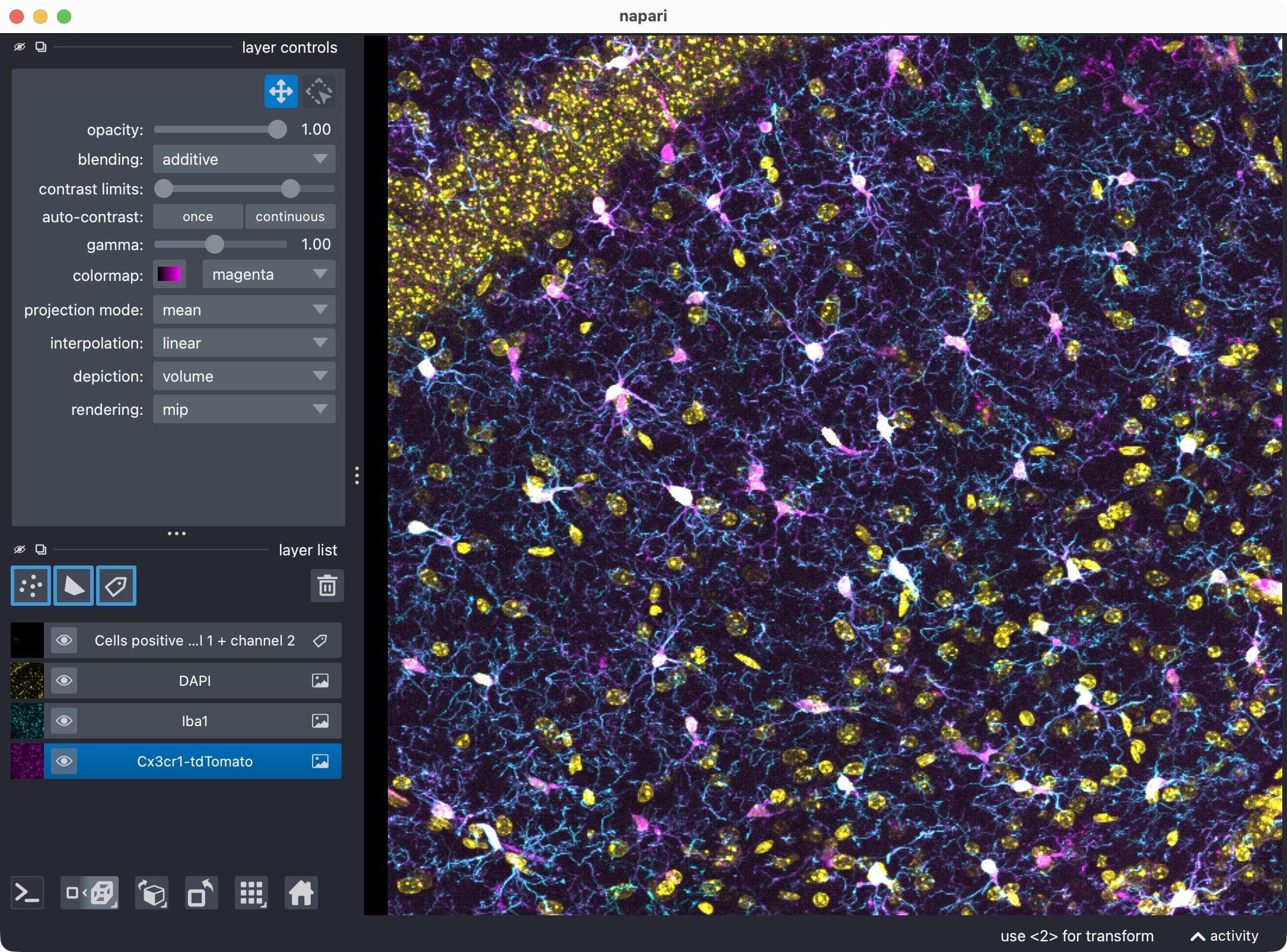
Top: Overview of the microglia (magenta), Iba1 (cyan), and optional third channel (yellow), along with the segmentation layer of the microglia cells that are positive for both marker channels.
Export results
The final cell writes the result bundle to the standardized results/
directory:
export_analysis_outputs(
run_result=run_result,
paths=loaded_images.paths,
optional_region_result=None)
print("Final results exported.")
As in the other tutorials, the export is intentionally placed at the end so that the saved outputs reflect the final accepted state after optional refinement or manual editing.
When to use this tutorial
This three-channel workflow is especially useful when:
channel 3 should contribute more than occupancy,
you want separate cell-positivity calls for two marker channels,
you want to visualize different positivity subsets explicitly,
you want to demonstrate or test CellColoc’s generalized third-channel logic.
For simpler projects, the 2D or standard 3D tutorials are usually the better starting point. This tutorial is best understood as the advanced extension of those workflows.